Ważenie zgodnie z Farmakopeą USA
Autor: Mettler Toledo
1 Wprowadzenie
Ważenie jest jedną z najczęściej wykonywanych czynności w laboratorium kontroli jakości. Zazwyczaj jest to jedno z pierwszych ogniw łańcucha analizy, na przykład kiedy próbka lub wzorzec są przygotowane do późniejszego rozcieńczenia i analizy HPLC lub qNMR. Ponieważ wszystkie błędy pomiaru masy mogą rzutować na cały proces analizy i powodować utratę precyzji w wynikach końcowych, Farmakopea Stanów Zjednoczonych (USP) określiła rygorystyczne wymagania dotyczące wag używanych do ważenia analitów w badaniach ilościowych. Wymagania te powinny gwarantować, że błędy ważenia w analizie będą małe lub nawet pomijalne. W tym opracowaniu technicznym szczegółowo omówiono wymagania dotyczące ważenia określone w Farmakopei Stanów Zjednoczonych oraz podano wskazówki, jak wdrożyć je w praktyce, aby zapewnić niezmiennie wysoką jakość wyników ważenia. Oprócz obowiązkowego rozdziału ogólnego 41 „Wagi”, który określa trzy kluczowe wymagania dla wag używanych do ważenia analitów w badaniach ilościowych, USP dostarcza szczegółowe wytyczne dotyczące najnowocześniejszej strategii kwalifikacji i eksploatacji wag w rozdziale ogólnym 1251 „Ważenie na wadze analitycznej”. Przedstawienie wszystkich informacji zawartych w rozdziale ogólnym 1251 wykraczałoby poza zakres tego opracowania; ujęto w nim zagadnienia związane z rutynowymi testami urządzeń stosowanymi przez użytkowników, ponieważ mają one kluczowe znaczenie dla zapewnienia ciągłej pracy urządzenia zgodnie z wymaganiami i jego „zgodności z przeznaczeniem”.
2 Rola farmakopei
Najważniejszym dla przemysłu farmaceutycznego dokumentem jest farmakopea, czyli zbiór opublikowanych norm, które określają wymagania dotyczące testowania chemicznych lub biologicznych substancji farmakologicznych, formy dawkowania oraz metody analizy leków. Normy te ustanawia się w celu zagwarantowania właściwego składu, mocy, jakości, czystości i jednorodności produktów. Farmakopee opisują wymagania dotyczące testowania substancji farmakologicznych i metod dawkowania, jednak nie regulują produkcji.

Rysunek 1: Założenie konwencji Farmakopei Stanów Zjednoczonych dn. 1 stycznia 1820 roku w sali Capitolu (obecnych 11 z 16 delegatów — sami lekarze) [1]. Pierwsza USP została opublikowana 21 grudnia 1820 r. Artysta: Robert Thom. Obraz zamówiony przez USP, 1957.
W USA „Farmakopea Stanów Zjednoczonych” (USP) została opublikowana po raz pierwszy w roku 1820 i była jedną z pierwszych farmakopei na świecie. Dziś istnieje wiele międzynarodowych farmakopei, np. Międzynarodowa Farmakopea Światowej Organizacji Zdrowia (WHO), Farmakopea Europejska (Ph. Eur.) lub Farmakopea Japońska (JP). Farmakopea Amerykańska jest jednak często uznawana za standard de facto na świecie, a jej monografie i metody są regularnie stosowane przez inne farmakopee i stosowane w ponad 140 krajach. Przez lata rozwinęła się znacząca współpraca i wymiana naukowa pomiędzy poszczególnymi farmakopeami. Jeden z najważniejszych programów został zainicjowany przez trzy najbardziej wpływowe farmakopee (USP, Farmakopea Europejska i Farmakopea Japońska), które wdrożyły program mający na celu osiągnięcie zharmonizowanych standardów w trzech organizacjach w ramach formalnego, etapowego procesu (Grupa Dyskusji Farmaceutycznych PDG). W większości państw krajowe farmakopee są dokumentami o wiążącej mocy prawnej i opracowują je instytucje rządowe. USP nie jest jednak podmiotem rządowym, lecz radą powierniczą, którą wybiera konwencja USP. Decyzje naukowe i normalizacyjne podejmowane są przez radę ekspertów USP. USP de facto jest prawem opartym na mocy amerykańskiej federalnej ustawy o żywności, lekach i kosmetykach (Federal Food, Drug, and Cosmetics Act), która określa Farmakopeę Stanów Zjednoczonych i Krajowy Spis Leków (USP-NF) jako oficjalne kompendia leków sprzedawanych na rynku amerykańskim. Amerykańska Agencja ds. Żywności i Leków (FDA) egzekwuje stosowanie odpowiednich rozdziałów ogólnych i monografii podczas kontroli GMP. Sama USP nie odgrywa żadnej roli w egzekwowaniu prawa. USP-NF to połączenie dwóch kompendiów: Farmakopei Stanów Zjednoczonych (USP) i Krajowego Spisu Leków (NF). W USP omówiono monografie dotyczące substancji farmakologicznych, metod dawkowania i preparatów złożonych. Monografie dotyczące suplementów diety i składników diety znajdują się w odrębnej części USP. Monografie pomocnicze znajdują się w NF. USP-NF jest publikowane trzy razy w roku. Pełen tytuł zawiera numer wersji, który nie jest taki sam dla USP i NF. Na przykład w 2019 r. obowiązuje „USP 42-NF 37” czyli 42. Wersja USP i 37. wersja NF, a w 2020 r. obowiązywać będzie kompendium „USP 43-NF 38”. Numer wersji zmieniany jest odpowiednim suplementem dwa razy w roku.
Monografia obejmuje nazwę składnika lub preparatu, jego definicję, wymagania dotyczące pakowania, przechowywania i etykietowania oraz specyfikację. Specyfikacja obejmuje serie testów, procedury badań i kryteria akceptacji. Testy i procedury wymagają użycia oficjalnych wzorców referencyjnych USP. Składniki i produkty lecznicze muszą mieć określoną moc, jakość i czystość, aby były zgodne z wymaganiami monografii i odpowiednich rozdziałów ogólnych. Testy i procedury, ujęte w wielu monografiach, szczegółowo opisano w rozdziałach ogólnych USP-NF. Sekcja „Uwagi ogólne i wymagania” zawiera definicje terminów używanych w monografiach oraz informacje niezbędne do interpretacji wymagań monograficznych. Uwagi ogólne oraz rozdziały ogólne o numerach poniżej 1000 i powyżej 2000 opisują obowiązujące wymagania, natomiast rozdziały ogólne o numerach od 1000 do 1999 służą wyłącznie celom informacyjnym. Stanowią zbiór zasad najlepszej praktyki i są często uznawane lub wdrażane przez branżę, ale nie są prawnie egzekwowane przez amerykańską agencję FDA.
3 Wymagania dotyczące ważenia w USP
Najważniejsze informacje na temat ważenia znajdują się w rozdziale ogólnym 41 USP „Wagi” [2], który opisuje konkretne kryteria działania wag podczas wykonywania czynności związanych z kontrolą jakości w laboratorium. Aby zrozumieć dokładny zakres zastosowania rozdziału ogólnego 41, należy zapoznać się z uwagami ogólnymi i wymaganiami USP. Zawierają one dwa ważne paragrafy, które należy wziąć pod uwagę. W punkcie 6.50.20 „Rozwiązania” wskazano, że: “[…] Rozwiązania do pomiarów ilościowych należy przygotowywać z użyciem dokładnie zważonych lub dokładnie odmierzonych analitów (patrz punkt 8.20 Informacje)”. Część 8.20 „Informacje” zawiera następujące informacje: Część „Informacje” określa ilość do 10%. Jeśli wymagane jest „dokładne zmierzenie” lub „dokładne zważenie”, należy postępować zgodnie ze wskazaniami w częściach Urządzenia do pomiaru objętości 〈31〉 lub Wagi 〈41〉. Na podstawie tych dwóch wymogów widać, że kiedy monografia wymaga „dokładnego zważenia” materiału, waga używana do ważenia musi spełniać wymagania opisane w rozdziale ogólnym 41. Ponadto, zgodnie z zasadą dotyczącą wszystkich monografii, anality używane do pomiarów ilościowych muszą być ważone na wadze spełniającej wymagania rozdziału ogólnego 41. Należy ponownie stwierdzić, że USP i inne farmakopee określają tylko wymagania dotyczące testowania chemicznych i biologicznych substancji farmakologicznych oraz metod dozowania i analizy leków. Innymi słowy, nie mają one zastosowania do produkcji aktywnych składników farmaceutycznych, substancji pomocniczych ani gotowych farmaceutyków Rozdział ogólny 41 jest uzupełniony rozdziałem ogólnym 1251 „Ważenie na wadze analitycznej” [3]. Zawiera on dodatkowe informacje i najnowocześniejsze koncepcje dotyczące ważenia analitycznego. Jak wspomniano wcześniej, wszystkie rozdziały ogólne o numerach od 1000 do 1999 służą wyłącznie celom informacyjnym i nie są egzekwowane przez amerykańską agencję FDA podczas audytów GMP. Wiele firm wdraża te koncepcje dobrowolnie, aby osiągnąć wysoki poziom jakości, który można się pochwalić w materiałach dla kadry kierowniczej oraz podczas audytów wewnętrznych i zewnętrznych.
4 USP, rozdział ogólny 41 „Wagi”
4.1 Dokładność i precyzja
Zgodnie z tym, co pisaliśmy wcześniej, w Farmakopei Stanów Zjednoczonych (USP) znalazły się rygorystyczne wymogi dotyczące wag, które są używane do ważenia analitów przeznaczonych do badań ilościowych. Wymagania te powinny gwarantować, że błędy ważenia w analizie będą małe lub nawet pomijalne. W rozdziale ogólnym 41 określono trzy różne wymagania dotyczące materiałów, które wymagają dokładnego zważenia w celu osiągnięcia tego celu (zmieniono format cytatu, aby był bardziej zrozumiały). „Ważenie powinno być wykonywane przy użyciu wagi, która • przeszła wzorcowanie w zakresie roboczym i spełnia określone wymagania • powtarzalności • i dokładności pomiaru”. Zanim szczegółowo przeanalizujemy trzy wymagania dotyczące wzorcowania, powtarzalności i dokładności, ważne jest, aby zrozumieć, jak w USP definiuje się dokładność. USP definiuje dokładność w rozdziale ogólnym 1225 „Walidacja procedur uzupełniających” jako [4]: „Dokładność procedury analitycznej polega na zbliżeniu wyników testów uzyskanych w tej procedurze do wartości rzeczywistej. Dokładność procedury analitycznej powinna być ustalana w całym jej zakresie. [Uwaga na temat terminologii: Definicja dokładności w tym rozdziale oraz w ICH Q2 dotyczy wyłącznie braku odchylenia. W międzynarodowym słowniku metrologii (VIM) i dokumentacji Międzynarodowej Organizacji Normalizacyjnej (ISO) „dokładność” ma inne znaczenie. W ISO koncepcja dokładności łączy brak odchylenia („poprawności”) z precyzją.] [5, 6, 7] Innymi słowy wymóg dotyczący dokładności opisany w rozdziale ogólnym 41 USP określa test, który ocenia systematyczne odchylenie wagi. We wszystkich innych dziedzinach metrologii pojęcie to nazywa się „poprawnością”. Z drugiej strony test powtarzalności pozwala ocenić precyzję wagi. Precyzja jest określana w podobny sposób w przemyśle farmaceutycznym oraz w VIM / ISO. W rozdziale ogólnym 1225 USP określono wymagania dotyczące precyzji: „Precyzja procedury analitycznej jest stopniem zgodności pomiędzy poszczególnymi wynikami badań przy wielokrotnym stosowaniu procedury […]. Precyzja procedury analitycznej jest zwykle wyrażana jako odchylenie standardowe lub względne odchylenie standardowe (współczynnik zmienności) serii pomiarów. […]”.

Innymi słowy, dwa testy określone w rozdziale ogólnym 41 USP mają na celu ocenę losowego i systematycznego błędu wagi. Określając odpowiednie kryteria zaliczenia dla obu testów, można kontrolować błędy losowe i systematyczne urządzenia. Jak ustalimy później, oba kryteria zaliczenia są wyrażone jako względne wartości graniczne 0,10%. Z praktycznego punktu widzenia wymóg ten jest dość rygorystyczny i zapewnia, że błąd pomiaru masy jest zwykle mały, a nawet pomijalny, w porównaniu z błędami na kolejnych etapach procesu opisanych w poszczególnych monografiach podczas testowania substancji farmaceutycznych (np. rozcieńczanie, analiza HPLC itp.). Należy zauważyć, że znaczenie pojęcia „dokładne ważenie” różni się od powyższej definicji „dokładności”: „dokładne ważenie” uwzględnia wszystkie trzy wymagania, wagę wzorcowaną oraz wagę, która spełnia określone wymagania dotyczące błędu losowego („precyzja”) i błędu systematycznego („dokładność”).
4.2 Wzorcowanie
Wzorcowanie to jedna z najważniejszych czynności, które należy wykonywać okresowo, jeśli urządzenia są używane do badania jakości. Istnieje wiele norm międzynarodowych, które regulują ten wymóg, np. ISO 9001 oraz zasady Good Laboratory Practice (GLP) i Good Manufacturing Practice (GMP). Niemal każda osoba pracująca w dziale kontroli jakości (QC) i zapewniania jakości (QA) — zarówno w laboratorium, jak i w środowisku produkcyjnym — zna stosowne wymagania określone w tych dokumentach. Nie ma jednak wspólnej interpretacji definicji wzorcowania, realizacji wzorcowania ani poszczególnych działań składających się na wzorcowanie. Dlatego proponujemy sprowadzić to pojęcie do wspólnego mianownika. a. Dokładnie, ale nie precyzyjnie b. Precyzyjnie, lecz nie dokładnie Rysunek 2: a) Wyniki, które są dokładne (prawdziwe), ale nieprecyzyjne (niepowtarzalne), wykazują szeroki rozrzut, ale wokół punktu centralnego; b) wyniki, które są precyzyjne, ale niedokładne (nieprawdziwe), pokazują ciasne zgrupowanie, ale nie wokół punktu centralnego. Wzorcowanie to zestaw czynności wykonywanych na urządzeniu pomiarowym w celu zrozumienia jego działania. Wykonuje się je poprzez ustalenie zależności między znanymi wartościami (wzorcami pomiarowymi) i powiązanymi wartościami pomiarowymi (wskazaniami) wzorcowanego urządzenia. Stosunek tych wartości jest obarczony odchyleniami i powiązaną z nimi niepewnością pomiaru. Międzynarodowy słownik metrologii (VIM) określa następującą definicję wzorcowania: „Działanie, które — w określonych warunkach— najpierw ustala zależność pomiędzy wartościami miary odwzorowanymi przez wzorce pomiarowe a odpowiadającymi im wskazaniami wraz z niepewnością pomiaru, a w drugim kroku wykorzystuje tę informację do ustalenia zależności pozwalającej uzyskać wynik pomiaru na podstawie wskazania”. Widać, że związek pomiędzy wartościami znanymi i zmierzonymi można ustalić tylko wtedy, gdy uzyskane zostaną powiązane z nimi niepewności pomiaru. Niestety w praktyce powszechnie panuje błędne zrozumienia wzorcowania: wielu użytkowników spoza laboratorium kalibracyjnego nie uwzględnia niepewności pomiaru podczas „wzorcowania” przyrządu. Niepewność pomiaru jest zdefiniowana w przewodniku „Guide to the Expression of Uncertainty in Measurement” (GUM) [8] jako: „Parametr związany z wynikiem pomiaru, charakteryzujący rozrzut wartości, którą można przypisać do mierzonej zmiennej”. Zasadniczo niepewność pomiaru określa, jak daleko wynik pomiaru może odbiegać od wartości rzeczywistej z pewnym prawdopodobieństwem. Wyniki pomiaru i związana z nimi niepewność są dokumentowane w raportach wzorcowania lub certyfikatach ce. Jeśli „wzorce pomiarowe” używane w procesie wzorcowania są metrologicznie identyfikowalne (najlepiej w jednostkach SI), a niepewność pomiaru jest szacowana prawidłowo, wzorcowanie zapewnia metrologiczną identyfikowalność wyników pomiaru uzyskanych na wzorcowanym urządzeniu, pod warunkiem że laboratorium korzysta z ogólnie przyjętych metod.
Norma ISO/ IEC 17025 [9] określa ogólne wymagania dotyczące kompetencji laboratoriów w przeprowadzaniu testów i/lub wzorcowania. Akredytacja organu akredytacyjnego to formalny proces potwierdzający, że laboratorium jest kompetentne i spełnia wymagania określone w normie ISO/IEC 17025. Należy zauważyć, żena tym etapie istnieje błędne przekonanie co do istoty wzorcowania wykonywanego przez akredytowanego dostawcę. Akredytacja przyznawana jest w stałym zakresie i dla określonych czynności w każdej dziedzinie wzorcowania. Akredytacja nie jest „carte blanche” uprawniającą do wykonania wszelkich wzorcowań i kalibracji dla użytkownika przyrządu, który wymaga serwisu akredytowanego laboratorium kalibracyjnego. Certyfikat wzorcowania wydany przez akredytowane laboratorium jest zazwyczaj akceptowany bez konieczności przeprowadzania audytu laboratorium. Wynika to z międzynarodowych, wielostronnych porozumień między organizacjami akredytacyjnymi, np. Porozumienie o wzajemnym uznawaniu ILAC MRA). ILAC to międzynarodowa spółdzielnia akredytacji laboratoriów (International Laboratory Accreditation Cooperation). W odniesieniu do nieautomatycznych urządzeń wagowych (wag) istnieje wiele wytycznych dotyczących wzorcowania na poziomie krajowym, z których większość opiera się na koncepcjach opisanych w GUM. Choć wytyczne te są na ogół podobne, różnią się szczegółami, przez co opracowanie i stosowanie jednego podręcznika kalibracji na poziomie globalnym jest trudne lub wręcz niemożliwe. Społeczność naukowa podjęła w ostatnim czasie działania mające na celu rozwiązanie tego problemu poprzez stworzenie zharmonizowanej koncepcji wzorcowania nieautomatycznych urządzeń wagowych opartej na uznawanych na całym świecie wytycznych dotyczących wzorcowania. Procesem kieruje EURAMET, a opracowywany przez tę organizację podręcznik wzorcowania i kalibracji EURAMET cg-18 „Calibration of Non-Automatic Weighing Instruments” może zostać najpopularniejszym zbiorem tego rodzaju wytycznych na świecie [10]. W większości krajów europejskich jest podstawą akredytacji laboratoriów kalibracyjnych zgodnie z normą ISO/ IEC 17025 i został nawet przekonwertowany do systemu SIM (SIM jest ogólnoamerykańskim systemem metrologicznym), dzięki czemu jest bardzo często stosowany w obu Amerykach [11]. Jednak w Azji był do niedawna prawie nieznany. Wkrótce się to zmieni, ponieważ pierwsze laboratoria wzorcujące już otrzymały akredytację opartą na EURAMET cg-18 w Japonii, Tajlandii, Malezji, Singapurze, Indiach i Indonezji. Oczekuje się, że podręcznik ten zyska na popularności w Azji, dzięki czemu stanie się branżowym standardem stosowanym na poziomie globalnym. Wartość EURAMET cg-18 jest tym większa, że podręcznik ten nie tylko opisuje sposób ustalenia niepewności w czasie wzorcowania, ale także sposób szacowania tak zwanej niepewności wyniku ważenia, który charakteryzuje działanie urządzenia podczas codziennego użytkowania. Takie podejście ma duże znaczenie praktyczne, ponieważ ułatwia ocenę sprawności urządzeń względem wymagań dotyczących tolerancji ważenia określonych przez użytkownika. Na tym etapie istotne jest wyjaśnienie kolejnego błędnego myślenia o wzorcowaniu — oprócz wzorcowania przyrząd można również adiustować. VIM definiuje adiustację w następujący sposób: „zbiór czynności wykonywanych na systemie pomiarowym, tak aby jego wskazania odpowiadały wartościom mierzonej ilości”. Innymi słowy, podczas adiustacji urządzenia jego wskazania są modyfikowane w taki sposób, aby w jak największym stopniu odpowiadały wartościom ilościowym zastosowanych wzorców pomiarowych. Niestety wielu użytkowników stosuje słowa „wzorcowanie” i „adiustacja” zamiennie, niepoprawnie, a nawet przypadkowo [12]. Często mówi się o wzorcowaniu urządzenia wagowego, mając na myśli jego adiustację. VIM również zwraca na to uwagę słowami: „Adiustacji systemu pomiarowego nie należy mylić z wzorcowaniem, które jest warunkiem wstępnym adiustacji. Po adiustacji systemu pomiarowego, system pomiarowy wymaga zazwyczaj ponownego wzorcowania”. To stwierdzenie uwidacznia kolejny ważny aspekt wzorcowania: przed adiustacją urządzenia należy najpierw przeprowadzić wzorcowanie, aby zrozumieć i udokumentować jego zachowanie. Jest to szczególnie ważne, aby nie przerwać historii wzorcowania wcześniejszych pomiarów w urządzeniu. Po adiustacji przyrząd wymaga ponownego wzorcowania. Czasem użytkownicy mówią o wzorcowaniu przed modyfikacją (adiustacją) oraz o wzorcowaniu po każdej niezbędnej adiustacji i/lub naprawie. Podsumowując, rozdział ogólny 41 USP narzuca obowiązek wzorcowania wagi. Zazwyczaj wyniki wzorcowania są udokumentowane świadectwem wzorcowania wraz z szacunkową niepewnością pomiaru. Ten formalny proces jest wymagany zarówno z perspektywy zarządzania jakością, jak i zgodnie z normami ISO i GLP / GMP. Stanowi to podstawę nawet dwóch konkretnych ocen — powtarzalności i dokładności — opisanych w kolejnych rozdziałach.
4.3 Warunek powtarzalności — minimalna naważka
Zgodnie z USP, rozdział ogólny 41: „Powtarzalność jest oceniana przez zważenie jednego wzorca masy nie mniej niż 10 razy. […] Powtarzalność jest zadowalająca, jeśli dwukrotność odchylenia standardowego ważonej wartości podzielona przez najmniejszą pożądaną masę netto (tj. najmniejszą masę netto, jaką użytkownicy planują stosować na tej wadze) nie przekracza 0,10%. […]”. Ten wymóg definiuje konkretne badanie z określonym kryterium zaliczenia, które waga musi spełnić. W tym momencie ważne jest zrozumienie specyficznych zasad zaokrąglania obowiązujących we wszystkich rozdziałach USP. Zgodnie z sekcją „Uwagi ogólne i wymagania” punkt 7.20: „Obserwowane lub obliczone wartości zostaną zaokrąglone do takiej liczby miejsc po przecinku, która jest zgodna z wyrażeniem limitu. Liczby nie powinny być zaokrąglane, dopóki nie zostaną wykonane końcowe obliczenia wartości podlegającej zgłoszeniu. […]. Jeśli zaokrąglenie jest wymagane, należy uwzględnić tylko jedną cyfrę dziesiętną po prawej stronie ostatniego miejsca w wyrażeniu limitu. Jeżeli ta cyfra jest mniejsza niż 5, zostaje usunięta i poprzednia cyfra pozostaje niezmieniona. Jeżeli ta cyfra jest większa niż 5, zostaje usunięta, a poprzednia cyfra wzrasta o 1. Należy pamiętać, że w farmakopei USP termin „wyrażenie limitu” jest synonimem kryterium zaliczenia. Innymi słowy, do zaokrąglania zaobserwowanych lub obliczonych wartości do liczby miejsc po przecinku zgodnej z wyrażeniem limitu stosowane są matematyczne reguły zaokrąglania. Przykład: przy kryterium zaliczenia 0,10%, odchylenie standardowe 0,1049% zostanie zaokrąglone do 0,10%. Po osiągnięciu tej wartości waga zaliczy test powtarzalności. Odchylenie standardowe 0,1050% jest zaokrąglane do 0,11%, a waga nie zaliczy testu powtarzalności. Warto zwrócić uwagę, że procedura zaokrąglania znacznie różni się od zasad zaokrąglania we wszystkich innych dziedzinach metrologii stosowanej i legalizacyjnej, w których format limitu nie ma wpływu na zaokrąglenie wyniku pomiaru. Dolny limit powtarzalności 0,41d jest wyznaczony niezależnie od wyników testów, gdzie oznacza przedział wagi, nazywany również odczytem. To ograniczenie dotyczy błędu zaokrąglenia cyfrowego wskazania. W rozdziale ogólnym 1251 objaśniono dolną granicę odchylenia standardowego: „Dolny limit 0,41d dla wyników odchylenia standardowego wynika z błędu zaokrąglenia cyfrowego wskaźnika urządzenia wagowego. Błąd zaokrąglenia przypisany do pojedynczego odczytu jest obliczany jako 0,29d. Należy pamiętać, że ważenie zawsze składa się z dwóch odczytów: jednego przed umieszczeniem i drugiego po zdjęciu próbki z szalki, przy czym różnica pomiędzy tymi dwoma wskazaniami oznacza masę netto próbki. Dwa indywidualne błędy zaokrąglenia są zwykle dodawane po podniesieniu do 2. potęgi, prowadząc do wyniku 0,41d. Tarowanie urządzenia po umieszczeniu pojemnika tara na szalce nie ma wpływu na błądzaokrąglania, ponieważ wskaźnik zerowania jest również zaokrąglany. Bardzo ważną konsekwencją wymogu powtarzalności jest koncepcja minimalnej naważki, która została już opisana przed wieloma laty w rozdziale ogólnym 1251. Podczas niewielkiej zmiany rozdziału ogólnego 41 USP, która wchodzi w życie z dniem 1 sierpnia 2019 r., w tym rozdziale dodano opis minimalnej naważki. Chociaż test i ocena powtarzalności nie zostały w tej wersji zmienione, wprowadzono znaczenie minimalnej naważki dla praktycznego zastosowania wagi w codziennym użytkowaniu. Zgodnie z poprawionym rozdziałem ogólnym 41: „Pomiar powtarzalności określa najmniejszą masę netto materiału, jaką można zważyć na wadze, zgodnie z wartością graniczną 0,10%”. Przy odchyleniu standardowym i kryterium zaliczenia powtarzalności 2 × s ≤ 0,10% minimalna masa netto wszystkie masy równe lub większe niż 2 × s 0,10% spełniają niż te wymagania. Najmniejsza masa, która spełnia to kryterium, jest nazywana minimalną naważką: W wersji poprawionej rozdziału ogólnego 41 stwierdzono, że najmniejsze dopuszczalne odchylenie standardowe wynosi 0,41d, na podstawie zaokrąglenia wskazania cyfrowego. Dlatego najmniejsza możliwa minimalna naważka na wadze z odczytem d wynosi 2000 × 0,41d = 820d. Przykładowo w przypadku półmikrowagi o odczycie 0,01 mg najmniejsza dopuszczalna minimalna naważka wynosi 8,2 mg. Minimalna naważka jest zwykle wyższa od tego progu. mmin ≫ 820 × d Jeśli na przykład odchylenie standardowe dziesięciu powtórzonych pomiarów na półmikrowadze wynosi 0,007 mg, minimalna naważka zostanie obliczona jako 2000×0,007 mg=14 mg. Ten przykład pokazuje, że we wszystkich przypadkach minimalną naważkę należy określić metodą prób i błędów. Odchylenie standardowe zmienia się w zależności od miejsca instalacji i samego przyrządu. Dlatego nie można go uzyskać ze specyfikacji opublikowanych w arkuszach danych, a założenie, że minimalna naważka powinna wynosić 820d nie jest zasadne. mmin = 2 × s = 2000 × s 0,10% Jak określono w rozdziale „Warunek powtarzalności”, najmniejsza masa netto jest definiowana jako najmniejsza ilość substancji, którą użytkownik chce ważyć na urządzeniu w codziennej pracy. Najmniejsza masa netto to wymóg użytkownika i nie należy jej mylić z minimalną naważką, która jest właściwością urządzenia obliczaną zgodnie z powyższym opisem. W przypadku tych dwóch definicji zastosowanie ma następujące twierdzenie: Jeśli najmniejsza masa netto (którą użytkownik chce zważyć) jest równa lub większa od minimalnej naważki (obliczonej na podstawie powtarzalności wagi), kryterium powtarzalności z rozdziału ogólnego 41 jest spełnione. W tym momencie należy ponownie zaznaczyć, że podczas ważenia próbki nie należy uwzględniać masy naczynia wagowego. Jest to uwidocznione w jednym ze zdań rozdziału ogólnego 41, które wyraźnie odnosi się do ilości netto materiału: „Pomiar powtarzalności określa najmniejszą masę netto materiału, jaką można zważyć na wadze, zgodnie z wartością graniczną 0,10%”. Ponadto w rozdziale ogólnym 1251 wskazano: „Minimalna naważka odnosi się do masy próbki, a nie do masy tara ani do masy brutto”. Również w przypadku odważania różnych próbek do tego samego pojemnika tara masa każdej pojedynczej próbki musi być równa lub większa od minimalnej naważki. Omówimy teraz kolejne ważne stwierdzenie z części dotyczącej powtarzalności w rozdziale ogólnym 41: „Wzorzec masy musi się mieścić w zakresie roboczym wagi, ale nie wymaga wzorcowania. Ponieważ odchylenie standardowe jest praktycznie niezależne od masy próbki w zakresie ważenia, użycie małego wzorca masy, który może być trudny w obsłudze, nie jest wymagane”. Ważne jest, aby wyjaśnić, czym jest zakres roboczy wagi: Jest to zakres działania wagi, w którym obowiązują określone wymagania dotyczące jej działania. W tym przypadku jest to zakres działania wagi, w którym spełnione jest kryterium powtarzalności. Po uwzględnieniu pojęcia minimalnej naważki zakres roboczy wagi zaczyna się od minimalnej naważki, a kończy na górnej granicy zakresu pomiarowego, przy maksymalnym zakresie ważenia.
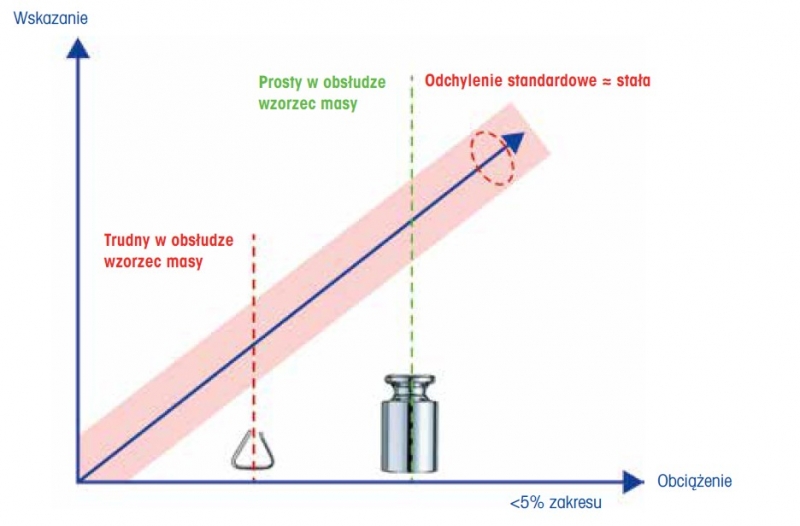
Rysunek 3: Do przeprowadzenia testów powtarzalności nie jest konieczne wybieranie wzorców masy w punkcie roboczym wagi. W rozdziale ogólnym 1251 USP wskazano, że wzorzec masy powinien wynosić zaledwie kilka procent zakresu ważenia wagi, np. blisko, ale poniżej 5% zakresu ważenia.
Istotną konsekwencją powyższego stwierdzenia jest fakt, że nie jest konieczny wybór obciążenia testowego w punkcie pracy wagi lub w jego pobliżu. Na przykład w przypadku półmikrowagi o oczekiwanej minimalnej naważce około 20 mg do testu powtarzalności nie jest potrzebny wzorzec masy 20 mg. Zgodnie z wymaganiami wzorzec masy powinien mieścić się w zakresie roboczym wagi, w tym przypadku od około 20 mg do maksymalnego zakresu ważenia. Rozdział ogólny 1251 USP sugeruje w tabeli 1, aby obciążenie testowe wynosiło kilka procent nominalnego zakresu ważenia wagi, np. 10 g dla wagi o znamionowym zakresie ważenia 200 g. Obciążenie testowe, które jest wystarczająco większe od oczekiwanej minimalnej naważki, jest łatwiejsze do przenoszenia i pozwala uniknąć błędów w przenoszeniu, które mogłyby mieć negatywny wpływ na ocenę działania urządzenia.
4.4 Warunek dokładności
Oprócz powtarzalności w rozdziale ogólnym 41 USP określono konkretne badanie z kryteriami zaliczenia dla dokładności. „Dokładność wagi jest zadowalająca, jeśli podczas testu przy użyciu odpowiednich wzorców masy jest w granicach 0,10% wartości wzorca masy. Wzorzec masy jest odpowiedni, jeśli ma masę od 5% do 100% zakresu ważenia wagi”. Ten test dokładności opisuje ocenę błędu systematycznego urządzenia. Należy zauważyć, że w dolnej granicy zakresu pomiarowego wagi analitycznej najbardziej istotnym czynnikiem wpływającym na błędy pomiaru masy jest powtarzalność. Z tego względu ocena dokładności za pomocą małych wzorców masy nie jest istotna, ponieważ potencjalny błąd systematyczny byłby w całości zamaskowany ograniczoną precyzją urządzenia, wyrażoną jako powtarzalność. Aby uniknąć używania małych obciążeń do testowania systematycznych odchyleń, w rozdziale ogólnym 41 USP określono, że masa obciążenia testowego powinna wynosić co najmniej 5% zakresu ważenia wagi. Aby spełnić wymagania dotyczące dokładności, wystarczy użyć jednego wzorca masy, co jest widoczne w sformułowaniu “[…] podczas testów z odpowiednim wzorcem (lub wzorcami) masy […]”. Ocena dokładności w całym zakresie pomiarowym urządzenia nie wymaga użycia wielu wzorców masy. Jak wynika z rozdziału ogólnego 41 USP, do oceny dokładności wagi wystarczy jedno obciążenie testowe. Jest to zgodne z podejściem opartym na ryzyku, które również zostało opisane w rozdziale ogólnym 1251, gdzi wskazano, że czynności służące kwalifikacji działania powinny uwzględniać znaczenie właściwości wagi dla działania urządzenia. Zazwyczaj to czułość jest najważniejszą cechą wagi przy ocenie błędu systematycznego, więc w praktyce w celu spełnienia wymagań określonych w rozdziale ogólnym 41 wykonywany jest tylko test czułości. Zgodnie z rozdziałem ogólnym 1251 typowe obciążenie testowe czułości znajduje się na poziomie lub dostatecznie blisko zakresu ważenia wagi.
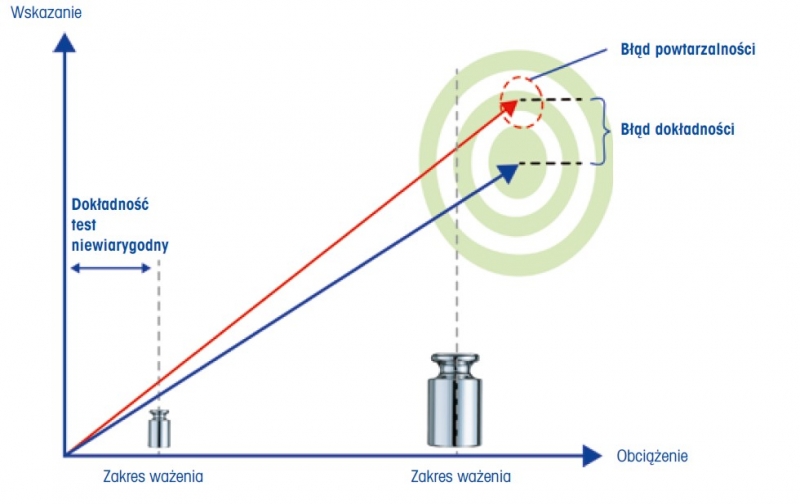
Rysunek 4: W celu przetestowania dokładności, wzorzec masy powinien mieścić się w zakresie od 5% do 100% zakresu ważenia.
W rozdziale ogólnym 41 USP określono także wymagania dotyczące dokładności obciążeń testowych wykorzystywanych podczas testu. „Maksymalny dopuszczalny błąd wzorca masy (mpe) lub jego niepewność wzorcowania nie powinna przekraczać jednej trzeciej zastosowanego limitu testu dokładności”. Jeśli wzorzec masy jest zgodny z wymogiem mpe (podanym w ważnym certyfikacie wzorcowania), do testu dokładności wystarczy uwzględnić nominalną wartość masy wzorców. Następnie użytkownik musi upewnić się, że maksymalny dopuszczalny błąd nie przekracza jednej trzeciej kryterium zaliczenia. W przeciwnym razie, jeśli zostanie uwzględniona wartość wzorcowanego obciążenia testowego (nominalna wartość masy podana w ważnym certyfikacie wzorcowania), niepewność wzorcowania nie powinna przekraczać jednej trzeciej kryterium zaliczenia. Jeśli kryterium zaliczenia jest ustawione na 0,05% zgodnie z informacjami zawartymi w rozdziale ogólnym 1251 USP, maksymalny dopuszczalny błąd lub niepewność wzorcowania wzorca masy nie może przekraczać 0,016%. Co ważne, kryterium zaliczenia 0,05% zostało wyjaśnione w rozdziale 5.1 Kwalifikacja działania. Wymagania dotyczące wzorców masy nie są zbyt rygorystyczne i w wielu przypadkach wystarczające są wzorce masy klasy OIML F1 lub F2 bądź też klasy ASTM 3 lub 4. Oto przykład: Mikrowaga o zakresie ważenia 20 g jest testowana za pomocą wzorca masy 20 g zgodnie z rozdziałem ogólnym 1251, tym samym wykorzystując jego nominalną wartość masy. Maksymalny dopuszczalny błąd tego wzorca masy 20 g nie powinien przekraczać 0,016% z 20 g, tj. 3,33 mg. Wzorzec masy 20 g klasy OIML F2 charakteryzuje się dokładnością ±0,8 mg (patrz OIML R111-1, tabela 1 [13]), więc wymagania w zakresie dokładności pomiaru masy określone w rozdziale 41 są spełnione z nadmiarem. 5 Rozdział ogólny 1251 USP „Ważenie na wadze analitycznej” Rozdział ogólny 1251 USP zawiera szczegółowe informacje dotyczące kwalifikacji i obsługi urządzenia. Rozdział ten jest poświęcony wagom analitycznym, ale większość przedstawionych informacji można również zastosować do wag o wyższym zakresie ważenia, takich jak wagi precyzyjne lub platformowe. W tym opracowaniu technicznym omówiono tylko dwa najważniejsze aspekty rozdziału ogólnego 1251 USP, czyli kwalifikację działania i współczynnik bezpieczeństwa.
5.1 Kwalifikacja działania wag
Zgodnie z rozdziałem ogólnym 1058 USP „Kwalifikacja urządzeń analitycznych” [14], kwalifikacja działania to „udokumentowany zbiór czynności niezbędnych do wykazania, że urządzenie zawsze działa zgodnie ze specyfikacją zdefiniowaną przez użytkownika i jest odpowiednie do zamierzonego zastosowania”. Z definicji wynika, że kwalifikacja działania nie jest czynnością jednorazową, ale należy ją przeprowadzać okresowo. Wzorcowanie to podstawowa czynność wykonywana podczas kwalifikacji działania, wymagana także w rozdziale ogólnym 41 USP. Rozdział ogólny 1251 USP nie zawiera jednak szczegółowych informacji o wzorcowaniu, lecz skupia się na pozostałych czynnościach wykonywanych zazwyczaj przez użytkownika pomiędzy wzorcowaniami, często nazywanych rutynowymi testami użytkownika lub po prostu rutynowymi testami. Zaleca się przeprowadzenie analizy ryzyka w celu oceny krytycznych warunków zastosowania, do którego waga jest przeznaczona. Na podstawie tej analizy ryzyka można określić typ i częstotliwość rutynowych testów. Czułość, liniowość, niecentryczność i powtarzalność są zwykle parametrami ważenia, które można uwzględnić w analizie ryzyka. Testowane są jednak zazwyczaj tylko te parametry ważenia, które mają znaczący wpływ na działanie urządzenia. W rozdziale ogólnym 1251 zamieszczono dalsze szczegóły dotyczące sugerowanych testów wydajności służących ocenie dokładności wagi: “Czułość, liniowość i niecentryczność wpływają na odchylenie systematyczne, tzn. ograniczają dokładność wagi […]. Ponieważ odchylenia są w dużej mierze niezależne od siebie, mało prawdopodobne jest, aby wszystkie odchylenia wystąpiły jednocześnie i posiadały ten sam znak algebraiczny. W związku z tym arytmetyczne zsumowanie wszystkich odchyleń w celu oceny dokładności wagi byłoby dość ostrożnym zabiegiem. Dodanie kwadratów poszczególnych odchyleń jest bardziej realistyczne. Dzięki alokacji 50% budżetu tolerancji ważenia na kryteria zaliczenia poszczególnych właściwości, np. czułości, liniowości i niecentryczności, analitycy zapewniają zgodność z wymaganą tolerancją ważenia. W związku z tym kryteria zaliczenia dla poszczególnych właściwości uwzględniających odchylenia systemowe ustala się jako tolerancję ważenia podzieloną przez 2. Własności te — lub ich podzbiór — mogą także spełnić warunek dokładności opisany w 〈41〉. W tym przypadku kryteria zaliczenia pozwalają więc na maksymalne odchylenie czułości, liniowości i niecentryczności o 0,05%”. To wyjaśnia, dlaczego warto ustawić kryterium zaliczenia testu czułości na 0,05% zamiast 0,1%, czyli zgodnie z rozdziałem ogólnym 41, po uwzględnieniu ogólnej dokładności urządzenia („suma wszystkich systematycznych odchyleń”). Z uwagi na znaczenie rutynowych testów w codziennej pracy urządzeń możemy dostarczyć dodatkowych informacji na temat testowania poszczególnych parametrów ważenia.
a. Powtarzalność
W przypadku laboratoryjnych urządzeń wagowych większość ważonych próbek, zwłaszcza do analizy ilościowej, odpowiada warunkowi „małej próbki”, tzn. próbki o masie netto znacznie mniejszej niż zakres ważenia urządzenia wagowego. Niepewność pomiaru urządzeń wagowych na dolnym końcu zakresu pomiarowego zależy od powtarzalności, patrz rysunek 4. W związku z tym w większości procesów ważenia powtarzalność jest najważniejszym czynnikiem wpływającym na niepewność i dlatego powinna byćokresowo testowana przez użytkownika. Powtarzalność najlepiej oceniać dla masy równej kilku procentom
maksymalnego zakresu ważenia. Zaleca się użycie kolejnego dostępnego pojedynczego wzorca masy zgodnie z klasyfikacją OIML lub ASTM, która jest mniejsza lub równa 5% zakresu ważenia urządzenia wagowego. Ponieważ powtarzalność jest zasadniczo stała dla małych mas próbek (patrz rysunek 3), można ją uznać za reprezentatywną dla całego dolnego zakresu ważenia. W krytycznych zastosowaniach wag analitycznych i mikrowag, gdzie są ważone małe ilości, zaleca się umieszczenie na szalce jako obciążenia wstępnego przedmiotu tara typowego dla danego zastosowania (pojemnika, kolby itp.), zwłaszcza jeśli ma on dużą powierzchnię. Przedmiot tara może zwiększyć powtarzalność ze względu na prądy powietrza i efekty konwekcji oddziałujące na jego powierzchnię [15], a test z realistyczną tarą daje bardziej realistyczny wynik testu powtarzalności.
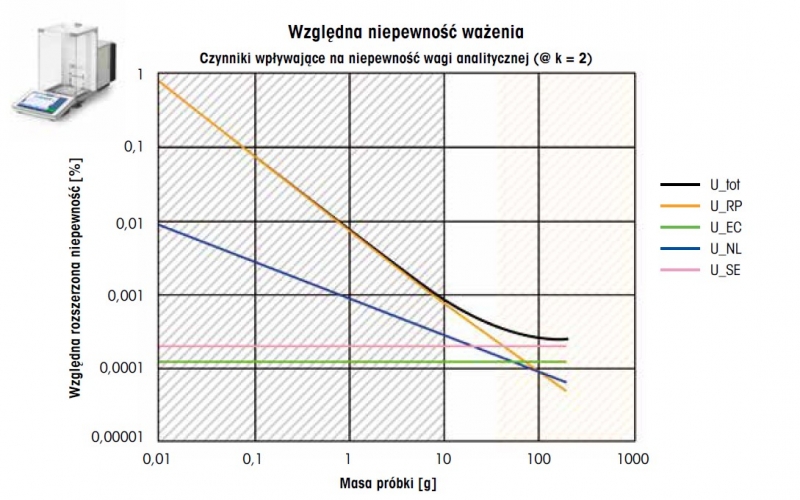
Rysunek 5: Względna rozszerzona niepewność pomiaru w stosunku do masy próbki na wadze analitycznej o zakresie ważenia 200 g (U_tot, gruba czarna krzywa). Przedstawiono również czynniki wpływające na niepewność: powtarzalność (U_RP, pomarańczowy), niecentryczność (U_EC, zielony), nieliniowość (U_NL, niebieski) i odchylenie czułości (U_SE, różowy). Niepewność zwiększa się o współczynnik k = 2. W obszarze zaznaczonym na szaro (przy niskiej masie próbki) powtarzalność ma dominujący wpływ na niepewność. Czułość i niecentryczność to główne czynniki występujące w obszarze zaznaczonym kolorem pomarańczowym (przy dużej masie próbki).
b. Czułość
Test czułości jest kolejnym ważnym testem, który użytkownik powinien wykonywać regularnie. Odchylenie czułości jest liniowe w stosunku do obciążenia i zwykle ogranicza dokładność urządzenia w górnej części zakresu ważenia, patrz rysunek 5. Dlatego test czułości jest wykonywany z masą bliską maksymalnemu zakresowi ważenia, najlepiej z jednym obciążeniem testowym. Zaleca się użycie kolejnego dostępnego pojedynczego wzorca masy zgodnie z klasyfikacją OIML lub ASTM [13, 16], która jest mniejsza lub równa nośności urządzenia wagowego. Nadal powszechną praktyką w wielu branżach jest testowanie urządzeń wagowych pod kątem odchyleń systematycznych w tzw. punkcie roboczym, czyli z obciążeniem odzwierciedlającym ilość ważonej substancji w typowym zastosowaniu. Ta praktyka jest odpowiednia dla punktów roboczych w górnej części zakresu pomiarowego, jednak nie jest odpowiednia, jeśli punkt roboczy znajduje się w dolnej części zakresu pomiaru ze względu na dominujący wpływ powtarzalności.
c. Niecentryczność
Na działanie urządzeń wagowych mogą również wpływać odchylenia wynikające z niecentryczności. Zazwyczaj odchylenia niecentryczności występują częściej w przypadku wag używanych w strefie produkcji ze względu na odmienne zasady konstrukcji, wysokie narażenie na naprężenia mechaniczne lub uszkodzenia oraz wyższe prawdopodobieństwo, że przedmiot zostanie umieszczony niewspółśrodkowo na platformie. Dlatego testy niecentryczności są ogólnie ważniejsze w strefie produkcji niż w laboratorium. Jeśli jednak test niecentryczności dotyczy danego zastosowania, można go przeprowadzić z takim samym wzorcem masy, jak test czułości.
d. Liniowość
Testy odchylenia liniowości mają zazwyczaj mniejsze znaczenie, ponieważ wpływ liniowości na niepewność ważenia jest niewielki w każdym modelu elektronicznych urządzeń wagowych, patrz rysunek 5. Liniowość jest zwykle testowana tylko wtedy, gdy urządzenie wagowe jest wzorcowane, a nie podczas rutynowych testów pomiędzy wzorcowaniami.
e. Użycie wbudowanych wzorców masy
Oprócz testowania wag przy użyciu zewnętrznych wzorców masy, w praktyce stosowana jest również adiustacja przy użyciu wbudowanych wzorców masy. Taka praktyka umożliwia zmniejszenie częstotliwości testów czułości przy użyciu zewnętrznych wzorców masy [3, 17, 18]. Na tym etapie należy skomentować często spotykane, lecz błędne przekonanie, które dominuje w branży od dziesięcioleci. Niemal każdy pracownik branży farmaceutycznej, który zajmuje się kontrolą jakości, mówi o „codziennym teście wagi”, który dotyczy zwykle dokładności (a nie powtarzalności). Ma to związek z przekonaniem kolejnych pokoleń osób pracujących z wagami, którzy uważają, że waga powinna być testowana codziennie. Jest to pozostałość po epoce wag mechanicznych, w której codzienna kontrola była zalecana ze względu na zużycie, rozerwanie i ścieranie najważniejszych elementów mechanicznych. Wymóg ten nie dotyczy jednak wag elektronicznych, a zwłaszcza w przypadku wag elektronicznych z wbudowanym wzorcem masy codzienny test czułości z zewnętrznym wzorcem masy jest przestarzałą praktyką. Rozdział ogólny 1251 USP oraz inne materiały referencyjne [17] koncentrują się teraz na podejściu do testowania opartym na ryzyku. Jeden z aktualnych dokumentów źródłowych [18] wyraźnie mówi: „Dawniej zależało nam na przeprowadzaniu codziennych kontroli, ale podobnie jak w przypadku wzorcowania, częstotliwość tych kontroli powinna być określana na podstawie ryzyka związanego z danym zastosowaniem”. Rozdział ogólny 1251 USP dopowiada: „Częstotliwość kontroli wagi zależy od ryzyka w danym zastosowaniu i wymaganej tolerancji ważenia”.
5.2 Uwzględnianie współczynnika bezpieczeństwa
Na działanie wagi wpływa wiele czynników (zmiany otoczenia, różni operatorzy, różne naczynia wagowe itp.), dlatego z czasem ulega ono zmianom. Okresowe testy przeprowadzane przez użytkownika są zalecane w rozdziale ogólnym 1251 USP w celu zapewnienia, że waga stale spełnia określone wymagania, tj. że „nadaje się do określonego celu”. Jednym z parametrów o szczególnej czułości jest powtarzalność, szczególnie w przypadku ważenia niewielkich ilości substancji na wagach analitycznych lub mikrowagach. Poza rutynowymi testami wykonywanymi przez użytkownika dopuszcza się stosowanie w każdym urządzeniu pomiarowym tzw. współczynnika bezpieczeństwa. W przypadku wag współczynnik bezpieczeństwa stosuje się, ważąc tylko taką ilość materiału, której masa jest odpowiednio większa od minimalnej naważki, określonej w konkretnym czasie i w danych warunkach otoczenia przez upoważnionego technika lub użytkownika. Minimalna naważka jest obliczana na podstawie powtarzalności, a powtarzalność jest szczególnie wrażliwa na zmiany środowiska i obsługę przez użytkownika, więc zmienia się z upływem czasu. Ustawiając minimalną masę netto na wartość odpowiednio większą od minimalnej naważki, określonej w konkretnym czasie i w danych warunkach otoczenia, można stwierdzić, że instrument jest odpowiedni do danego celu, niezależni od zmian w działaniu przyrządu. Innymi słowy, najmniejsza masa netto, jaką użytkownik chce ważyć podczas normalnego użytkowania wagi, powinna być na tyle duża, aby była ona wyższa od minimalnej naważki, jaką ustalono w danym czasie.
Zgodnie z USP, rozdział ogólny 1251: „Czynniki, które mogą mieć wpływ na powtarzalność pracy wagi:
1. Wydajność wagi, a tym samym minimalna naważka, może się zmieniać ze względu na zmieniające się warunki otoczenia.
2. Poszczególni operatorzy mogą ważyć na wadze w różny sposób, tzn. minimalna naważka ustalana przez różnych operatorów może się różnić.
3. Odchylenie standardowe dla określonej liczby pomiarów masy jest tylko przybliżonym odchyleniem standardowym, które nie jest znane.
4. Wyznaczenie minimalnej naważki za pomocą wzorca masy może nie być całkowicie reprezentatywne dla danego zastosowania.
5. Naczynie tara może mieć wpływ na minimalną naważkę ze względu na wzajemne oddziaływanie otoczenia na powierzchnię naczynia wagowego.
Z tego względu, gdy jest to możliwe, pomiar masy powinien być wykonywany na wartościach większych niż minimalna naważka, tj. pożądana najmniejsza masa netto, jaką użytkownicy planują stosować z tą wagą, powinna być większa niż minimalna naważka”.
Koncepcja ta została określona ilościowo za pomocą współczynnika bezpieczeństwa. W zastosowaniach opisanych w rozdziale ogólnym 41 USP, gdzie minimalna naważka jest obliczana na podstawie odchylenia standardowego, współczynnik bezpieczeństwa jest definiowany jako iloraz pomiędzy najmniejszą masą netto a minimalną naważką. Przykładowo, jeśli użytkownik chce zważyć 30 mg na półmikrowadze (tj. kiedy najmniejsza masa netto wynosi 30 mg), a minimalna naważka wynosi 15 mg, współczynnik bezpieczeństwa wynosi 2. W tym momencie należy zauważyć, że definicja minimalnej naważki zgodnie z rozdziałem ogólnym 41 USP oraz opisana powyżej koncepcja współczynnika bezpieczeństwa są specjalnymi scenariuszami mającymi zastosowanie tylko do zastosowań opisanych w tym rozdziale. Co do zasady minimalną naważkę można obliczyć na podstawie niepewności pomiaru przyrządu wagowego, która jest podana w certyfikacie wzorcowania. Podczas wzorcowania uwzględniane są wszystkie istotne czynniki wpływające na niepewność pomiaru, a powtarzalność jest tylko jedną z nich, chociaż w przypadku wag analitycznych i mikrowag w dolnej części zakresu pomiarowego jest ona najważniejsza. Innymi słowy, obliczenie minimalnej naważki na podstawie rozdziału ogólnego 41 USP jest przybliżeniem prawdziwej minimalnej naważki, którą można obliczyć na podstawie niepewności pomiaru urządzenia. Jednak w przypadku wag analitycznych i mikrowag używanych w ramach USP podejście uproszczone, określone w rozdziale ogólnym 41 USP, jest ważne ze względu na dominującą rolę powtarzalności w porównaniu z innymi czynnikami wpływającymi na niepewność. Dlatego należy podkreślić, że w przypadku zastosowań wykraczających poza zakres rozdziału ogólnego 41 USP, zwłaszcza gdy rozważane są wagi precyzyjne, nie należy obliczać minimalnej naważki na podstawie wzoru podanego w rozdziale ogólnym 41, lecz na podstawie niepewności pomiaru urządzenia. Szczegółowe omówienie tego tematu wykracza poza zakres niniejszego opracowania, więc odsyłamy do odpowiednich źródeł [19].
6 Podsumowanie
Dokładne ważenie ma kluczowe znaczenie w każdym zastosowaniu, które wpływa na jakość produktu. USP określiła w rozdziale ogólnym 41 jasne wymagania, które powinny zagwarantować, że żaden pomiar wykonany zgodnie z USP nie przyczyni się w istotny sposób do błędów analitycznych. Oprócz wymogu wzorcowania wagi, wprowadza się wymagania dotyczące powtarzalności i dokładności, które podlegają ocenie w swoim zakresie tolerancji (0,10%). Istotną konsekwencją testu powtarzalności jest możliwość obliczenia minimalnej naważki określającej najmniejszą ilość substancji, jaką należy zważyć, aby uzyskać zgodność z wymaganiami w zakresie tolerancji. Rozdział ogólny 1251 USP zawiera szczegółowe informacje na temat rutynowych testów wag z zastosowaniem podejścia opartego na szacowaniu ryzyka w odniesieniu do działań związanych z weryfikacją działania. Ponadto omawia zmienność parametrów wagi w czasie, co prowadzi do koncepcji współczynnika bezpieczeństwa. Zastosowanie wymogów rozdziału ogólnego 41 USP oraz najlepszych praktyk zawartych w rozdziale ogólnym 1251 gwarantuje, że użytkownik będzie mógł polegać na dokładności urządzenia przez długi czas.
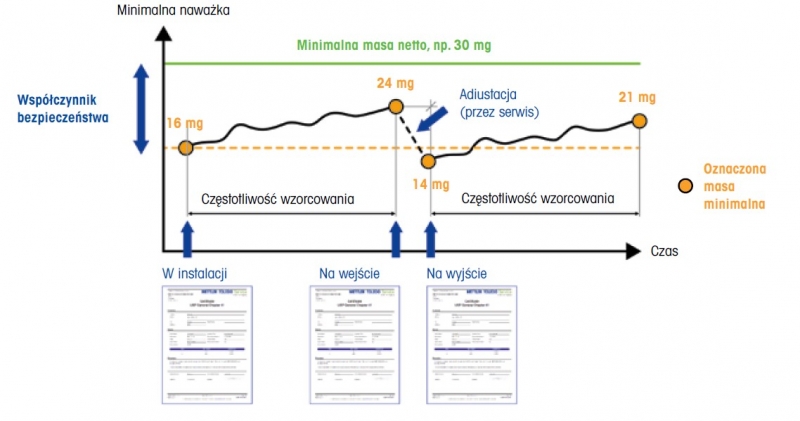
Rysunek 6: Zmienność masy minimalnej w czasie w zależności od zmieniających się warunków otoczenia. Pomarańczowe kropki oznaczają minimalne wartości masy uzyskane podczas określania powtarzalności. W celu zapewnienia, że najmniejsza masa netto jest zawsze większa niż minimalna naważka, należy zastosować współczynnik bezpieczeństwa. Aby zapewnić identyfikowalność wyników ważenia, przed serwisowaniem/ adiustacją urządzenia, należy przeprowadzić wzorcowanie „na wejście”.
Więcej informacji
Grupa METTLER TOLEDO
Dział Laboratoryjny
Kontakt: www.mt.com/contacts
Dane techniczne mogą ulec zmianie.
© 07/2019 METTLER TOLEDO. Wszelkie prawa zastrzeżone
30610004A
Group MarCom 2731 KR
7 Literatura
[1] G. Tirumalai, Long, A. “United States Pharmacopeial Convention: Respecting the past, moving confidently into the future”, United States Pharmacopeial Convention, Rockville, USA, pobrano ze strony www.usp.org.
[2] USP, rozdział ogólny 41 „Balances”, United States Pharmacopeial Convention, Rockville, USA.
[3] USP, rozdział ogólny 1251 „Weighing on an Analytical Balance”, United States Pharmacopeial Convention, Rockville, USA.
[4] USP, rozdział ogólny 1225 „Validation of Compendial Methods”, United States Pharmacopeial Convention, Rockville, USA
[5] JCGM 200 (VIM), “International Vocabulary of Metrology – Basic and General Concepts and Associated Terms”, 3. ed., JCGM, 2010.
[6] ISO 5725-1, “Accuracy (trueness and precision) of measurement methods and results – Part 1: General principles and definition”, International Organization for Standardization, Genewa, 1994.
[7] ICH Q2 (R1), “Validation of Analytical Procedures: Text and Methodology”, International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, 2005.
[8] JCGM 100 (GUM), “Evaluation of Measurement Data – Guide to the Expression of Uncertainty in Measurement”, JCGM, 2008.
[9] ISO/ IEC 17025, “General Requirements for the Competence of Calibration and Testing Laboratories”, International Organization for Standardization, Genewa, 2017.
[10] EURAMET cg-18, “Guidelines on the Calibration of Non-automatic Weighing Instruments”, 4. ed.,
EURAMET, 2015.
[11] SIM, “Guidelines on the Calibration of Non-automatic Weighing Instruments”, SIM, 2008.
[12] “Calibration of Weighing Instruments – Weighing in the Safe Weighing Range”, opracowanie techniczne, METTLER TOLEDO, 30260956, 2015.
[13] OIML R111-1, “Weights of Classes E1, E2, F1, F2, M1, M1–2, M2, M2–3 and M3, Part 1: Metrological and TechnicalRequirements”, OIML, 2004.
[14] USP rozdział ogólny 1058 “Analytical Instrument Qualification”, United States Pharmacopeial Convention,Rockville, USA.
[15] A. Reichmuth, “Weighing Small Samples on Laboratory Balances”, Proceedings of the 13th InternationalMetrology Congress, Lille, France, 2007.
[16] ASTM E617, “Standard Specifications for Laboratory Weights and Precision Mass Standards”, ASTM,Conshohoken, USA, 2018.
[17] Questions and Answers to Current Good Manufacturing Practices, Good Guidance Practices, Level 2Guidance – Equipment, U.S. Food and Drug Administration, Rockville, USA, pobrano ze stronyhttp://www.fda.gov/drugs/guidancecomplianceregulatoryinformation/guidanc…
[18] LAB 14, “In-house Calibration and Use of Weighing Machines”, 5. ed., United Kingdom Accreditation ServiceUKAS, Feltham, UK, 2015.
[19] “Safe Weighing Range Ensures Accurate Results”, METTLER TOLEDO, 30413341B, 2018