Dobra praktyka mikrobiologiczna w laboratorium w wytwórni farmaceutycznej
Autor: Jadwiga Marczewska, Krystyna Mysłowska
Laboratorium mikrobiologiczne w wytwórni farmaceutycznej podlega takim samym wymaganiom jak inne obszary działań w produkcji preparatów leczniczych, zgodnie z GMP (ang. Good Manufacturing Practice) [1, 2] lub innymi wymaganiami np.: 21 CFR 211 (ang. Code of Federal Regulations) [3], nawet jeśli owe wymagania ogólne są wzbogacone o wymagania specyficzne [4].
Warunki badań w laboratorium mikrobiologicznym
Aby zagwarantować niezawodność i odtwarzalność każdej operacji wykonywanej w laboratorium kontroli jakości konieczne jest wprowadzenie systemu zapewnienia jakości, którego główne zasady dotyczą również laboratorium mikrobiologicznego. Oceną jakości mikrobiologicznej w laboratorium kontroli jakości produktów leczniczych są objęte surowce i materiały opakowaniowe, woda do celów produkcyjnych i badawczych, półprodukty i produkty gotowe oraz warunki środowiska zarówno produkcyjnego jak i badań. Wykonywane są badania: jałowości, czystości mikrobiologicznej, obecności endotoksyn bakteryjnych, a także mikrobiologiczne badania witamin, oznaczanie aktywności antybiotyków, skuteczności ochrony przeciwdrobnoustrojowej oraz skuteczności preparatów dezynfekcyjnych.
|
Laboratorium mikrobiologiczne bierze udział w badaniach walidacyjnych dotyczących procesu wytwarzania oraz analiz badawczych. Udział laboratorium mikrobiologicznego jest niezbędny w badaniach stabilności substancji czynnych i gotowych produktów leczniczych. |
Wymagania ogólne dotyczące laboratoriów kontroli jakości w firmach farmaceutycznych są przedstawione w Rozporządzeniu Ministra Zdrowia w Części I w Rozdziale 6 Kontrola Jakości [2] i zostały omówione w artykule pt. „GMP w przemyśle farmaceutycznym” zamieszczonym w LAB 3/2013[5]. Wskazówki w zakresie działania laboratoriów mikrobiologicznych zawiera dokument EA-04/10 [6], który jest uzupełnieniem normy EN ISO/IEC 17025 [7]. Wytyczne zawarte w tym dokumencie mogą być również przydatne dla podmiotów pracujących zgodnie z GMP.
Zgodnie z Rozporządzeniem Ministra Zdrowia [2] pomieszczenia laboratorium muszą być odpowiednio zaprojektowane i zbudowane, wyposażone w urządzenia i sprzęt odpowiedni do prowadzonych operacji. Powinny być wystarczająco duże, aby uniknąć pomyłek i zanieczyszczeń. Wielkość pomieszczeń laboratoryjnych powinna być dostosowana do ilości wykonywanych analiz i ogólnej, wewnętrznej organizacji laboratorium. Typowe laboratorium mikrobiologiczne składa się z pomieszczeń badawczych, gdzie wykonuje się określone analizy mikrobiologiczne oraz pomieszczeń pomocniczych. Dobrą praktyką jest posiadanie odrębnych pomieszczeń do: odbioru, przechowywania, badania próbek w zakresie czystości mikrobiologicznej, badania próbek jałowych, inkubacji, przesiewów i badań diagnostycznych, a także przygotowania i sterylizacji pożywek hodowlanych i sprzętu, mycia i przechowywania szkła laboratoryjnego i sprzętu, prowadzenia sterylizacji, odkażania i niszczenia kultur bakteryjnych. Pomieszczenia dodatkowe stanowią: wejścia, korytarze, klatki schodowe, windy towarowe lub osobowe, pomieszczenia administracyjne (biura, archiwa) toalety i szatnie oraz magazyny.
W laboratoriach mikrobiologicznych, w których jest prowadzona kontrola produktów leczniczych, należy zagwarantować takie warunki badań, które są zgodne z wymaganymi obowiązującymi przy ich wytwarzaniu. Badania leków, dla których jest wymagana jałowość należy przeprowadzać w klasie czystości powietrza A w otoczeniu klasy czystości powietrza B [8]. Produkty lecznicze, w których badany jest parametr czystości mikrobiologicznej zwykle są badane w pomieszczeniach wyposażonych w boksy laminarne lub w szafy laminarne, zwane również komorami bezpiecznej pracy, w otoczeniu powietrza, dla którego wymagana czystość mikrobiologiczna jest taka, jak dla klasy czystości powietrza D, lub też w powietrzu o znanej i kontrolowanej liczbie zanieczyszczeń mikrobiologicznych. Wytwórca sam określa warunki w jakich wytwarza i bada niejałowe produkty lecznicze, ponieważ brak jest jednoznacznie określonych wymagań dla parametru czystości powietrza przy wytwarzaniu leków, dla których obowiązuje wymóg czystości mikrobiologicznej. Należy przyjąć, że warunki wytwarzania i kontroli dla tej grupy produktów leczniczych muszą zapewnić ich jakość zgodną z wymaganiami i dokumentacją dopuszczającą produkt do obrotu.
Laboratorium odpowiada za kilka krytycznych operacji, dzięki którym proces wytwarzania a następnie produkty gotowe spełniają ustalone parametry warunkujące ich zwolnienie do sprzedaży, bowiem gotowe produkty lecznicze muszą posiadać odpowiednie cechy jakościowe i być stabilne. Sposób, w jaki laboratorium realizuje te zadania, musi odpowiadać wewnętrznym i zewnętrznym normom jakościowym oraz procedurom.
Należy stworzyć takie warunki badań, które zapewnią izolację i ilościowe oznaczenie tylko tych drobnoustrojów, które mogą być obecne w próbach pobranych do analizy. Nie mogą być one przyczyną zanieczyszczenia środowiska przez drobnoustroje. W konsekwencji, nadzór nad warunkami badań powinien gwarantować wiarygodność uzyskiwanych wyników.
Ważnym elementem przy projektowaniu pomieszczeń laboratorium mikrobiologicznego jest przestrzeganie wymagań w zakresie bezpieczeństwa, które jest zależne od typu drobnoustrojów, z którymi pracujemy w laboratorium lub które spodziewamy się wykryć. Ze względu na fakt, że nie ma bezpiecznych mikroorganizmów istnieje ryzyko mikrobiologiczne, pośrednie lub bezpośrednie dla personelu i/lub otoczenia. Skutki ich działania mogą być funkcją liczby komórek mikroorganizmów, warunków środowiska oraz wrażliwości pracowników [9, 10].
Drobnoustroje są zakwalifikowane do czterech kategorii ryzyka, co jednocześnie warunkuje zastosowanie w laboratorium odpowiednich zabezpieczeń oraz procedur czyszczenia i dezynfekcji [11, 12]. Zgodnie z dyrektywą unijną 2000/54/WE i Rozporządzeniem Ministra Zdrowia z dnia 22 kwietnia 2005 r. jedynym kryterium określenia grupy ryzyka (zagrożenia) czynników biologicznych jest zdolność wywołania chorób zakaźnych [13].
Tabela 1. Klasyfikacja szkodliwych czynników biologicznych
|
Grupa 1. zagrożenia |
Czynniki, przez które wywołanie chorób u ludzi jest mało prawdopodobne. |
|
Grupa 2. zagrożenia |
Czynniki, które mogą wywołać choroby u ludzi, mogą być niebezpieczne dla pracowników, ale rozprzestrzenienie ich w populacji ludzkiej jest mało prawdopodobne. Zazwyczaj istnieją w stosunku do ich skuteczne metody profilaktyki lub leczenia. |
|
Grupa 3. zagrożenia |
Czynniki, które mogą wywołać u ludzi ciężkie choroby, są niebezpieczne dla pracowników, a rozprzestrzenienie czynników w populacji ludzkiej jest bardzo prawdopodobne. Zazwyczaj istnieją w stosunku do nich skuteczne metody profilaktyki lub leczenia. |
|
Grupa 4. zagrożenia |
Czynniki, wywołują u ludzi ciężkie choroby, są niebezpieczne dla pracowników, a rozprzestrzenienie czynników w populacji ludzkiej jest bardzo prawdopodobne. Zazwyczaj nie istnieją w stosunku do nich skuteczne metody profilaktyki lub leczenia. |
Pomieszczenia laboratorium powinny być zaprojektowane i wykonane zgodnie zasadą drogi jednokierunkowej „bez zawracania”, umożliwiające rozdzielenie wykonywanych czynności w czasie i przestrzeni. Przykładowy rozkład pomieszczeń laboratorium przedstawia rys. 1 [14]. Zasadą obowiązującą przy opracowaniu planu laboratorium mikrobiologicznego powinno być wykluczenie możliwości uzyskiwania fałszywych wyników z powodu niekontrolowanych wpływów czynników zewnętrznych.
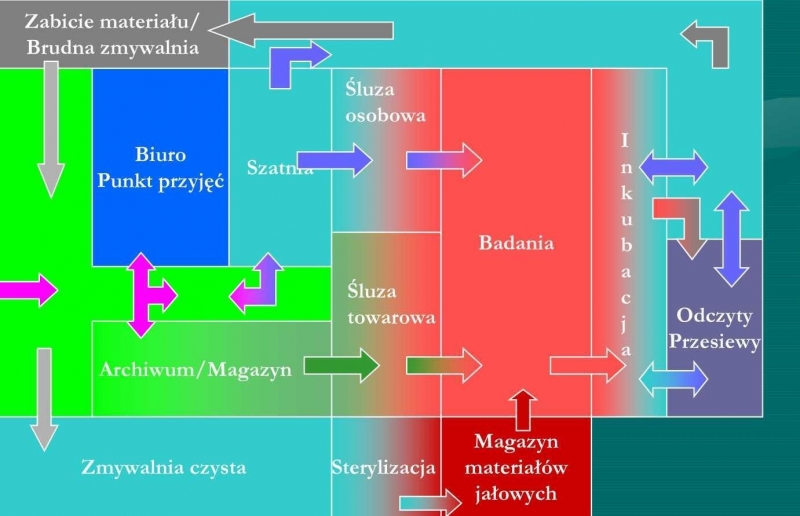
Rys. 1. Przykładowy rozkład pomieszczeń laboratorium
Reżimy higieniczne i parametry środowiska pracy
Aby zagwarantować odpowiednie reżimy higieniczne do przeprowadzania badań produktów leczniczych, w pomieszczeniach laboratorium mikrobiologicznego ściany, sufity i podłogi oraz meble powinny być gładkie, łatwe do mycia i dezynfekcji. Meble laboratoryjne powinny być tak zaprojektowane, aby ułatwić czyszczenie podłóg. Zaleca się wyposażyć laboratorium w meble ruchome, łatwe do przesuwania lub na wysokich nóżkach. Szafki należy zawieszać w taki sposób, aby sięgały do sufitu, co zabezpiecza przed zbieraniem się kurzu. Powietrze o odpowiedniej jakości, zgodnej z wymaganiami dla wykonywanych badań, należy wprowadzać poprzez system wentylacji połączony z filtrami dla powietrza wchodzącego i wychodzącego. Zalecana temperatura otoczenia w laboratorium to 18oC – 27oC. Aby chronić środowisko laboratorium przed szkodliwym oddziaływaniem promieniowania słonecznego należy zainstalować zewnętrzne żaluzje lub inne zabezpieczenia przeciwsłoneczne. Wewnętrzne instalowanie zabezpieczeń jest nieodpowiednie ze względu na możliwość gromadzenia się kurzu i powstawanie miejsc trudnych do wyczyszczenia i dezynfekcji.
W laboratorium ocenę jakości mikrobiologicznej badanych prób należy prowadzić w taki sposób, aby uniknąć zewnętrznego zanieczyszczenia mikrobiologicznego produktu badanego. Podejmowane środki ostrożności zabezpieczające przed zanieczyszczeniem nie mogą mieć wpływu na drobnoustroje, które mają być oznaczone w badaniu [15]. W celu zapewnienia odpowiedniej higieny środowiska i jego kontroli należy opracować program monitorowania środowiska i wskazać metody badań oraz określić poziom dopuszczalnych zanieczyszczeń mikrobiologicznych. Powinny być opracowane procedury postępowania w przypadku przekroczenia limitów zanieczyszczeń. Uzyskiwane wyniki powinny być analizowane w celu określenia trendów.
Personel i higiena na stanowiskach pracy
W laboratorium mikrobiologicznym powinna być zatrudniona wystarczająca liczba pracowników o odpowiednim wykształceniu i doświadczeniu. Personel zatrudniony w laboratorium mikrobiologicznym powinien przejść odpowiednie szkolenia umożliwiające mu prawidłową realizację powierzonych zadań [12]. Kierownictwo laboratorium powinno sformułować cele dotyczące wykształcenia, szkolenia i umiejętności personelu laboratorium. Zatrudnione osoby powinny mieć szeroką wiedzę i wystarczające doświadczenie w zakresie metod analiz mikrobiologicznych, potrafić ustalić dokładność i precyzję wymaganą do uzyskania akceptowalnych wyników. Zaleca się aby sprawdzanie kompetencji personelu było wykonywane regularnie na podstawie wyników uzyskiwanych w badaniach biegłości oraz podczas wewnętrznych sprawdzeń jakości badań. Należy prowadzić rejestr odbytych szkoleń i zdobytych kwalifikacji personelu wykonującego badania.
Zagrożenia ze strony personelu
Personel laboratorium może stanowić istotne źródło zanieczyszczenia próby. Dlatego też jest zobowiązany do zgłaszania każdego problemu zdrowotnego, jaki może mieć wpływ na wynik badania. Ważne jest aby w czasie wykonywania czynności z próbami, kulturami drobnoustrojów, pożywkami oraz podczas wykonywania posiewów unikać kaszlu i prowadzenia rozmów oraz innych czynności, które mogłyby mieć wpływ na badaną próbę. Zabronione jest ustne pipetowanie, spożywanie posiłków, picie jak również przechowywanie żywności w laboratoryjnych chłodziarkach i zamrażarkach.
Aby uniknąć zanieczyszczenia badanych próbek i pożywek, oraz ryzyka zakażenia personelu zatrudnionego w laboratorium, należy opracować odpowiednie procedury dotyczące higieny osobistej i higieny na stanowisku pracy:
- spożywania posiłków, picia i palenia,
- zachowania w pomieszczeniach badawczych,
- utrzymania czystości,
- ruchu osobowego w laboratorium (pracowników, gości, służb serwisowych),
- sposobu postępowania z jałową odzieżą ochronną
oraz instrukcje między innymi takie jak np.:
- mycia i odkażania rąk,
- noszenia i częstotliwości wymiany odzieży ochronnej,
- zachowania na wypadek wystąpienia nieoczekiwanego zdarzenia – pożar, porażenie prądem, przerwa w zasilaniu elektrycznym uwolnienie materiału zakażonego, skaleczenie lub choroba.
Należy przestrzegać podstawowych zasad higieny: nie przechowywać rzeczy osobistych oraz roślin w obszarze pracy laboratorium, nosić odzież ochronną odpowiednią do rodzaju wykonywanych badań.
Zaleca się zainstalować i korzystać z odpowiednich urządzeń do mycia rąk.
System dokumentacji i wymagania dotyczące urządzeń
Od laboratorium wymaga się, aby funkcjonował udokumentowany program dotyczący konserwacji, wzorcowana i sprawdzania parametrów wyposażenia oraz urządzeń wykorzystywanych do badań [8]. Zgodnie z dobrą praktyką laboratoryjną zaleca się, aby wyposażenie i urządzenia były utrzymywane w czystości i sprawności. Gdy to konieczne wyposażenie i przyrządy monitorujące należy wzorcować, tak aby zapewnić spójność pomiarową z odpowiednimi wzorcami. Urządzenia powinny być sprawdzane i serwisowane w celu zapewnienia bezpieczeństwa i dokładności wymaganej dla otrzymywanych wyników [12]. Rozporządzenie Ministra Zdrowia [2] w Rozdziale 3, pkt. 3.41 nakłada na wytwórców produktów leczniczych obowiązek kalibrowania i sprawdzania urządzeń do mierzenia, ważenia, rejestracji i kontroli, odpowiednimi metodami w określonych odstępach czasu. Zapisy dotyczące tych czynności należy przechowywać.
Aparatura i urządzenia powinny być tak skonstruowane i w taki sposób zainstalowane aby ułatwić wykonywanie czynności pomiarowych oraz umożliwić wygodną ich obsługę, wzorcowanie, czyszczenie i dezynfekcję.
W urządzeniach z kontrolą temperatury należy monitorować jej stabilność i równomierny rozkład. Gdy temperatura ma bezpośredni wpływ na wynik analizy lub ma zasadnicze znaczenie dla prawidłowej pracy urządzenia, przyrządy do pomiaru temperatury powinny być takiej jakości, aby gwarantowały wymaganą dokładność. W cieplarkach, łaźniach wodnych, sterylizatorach i pomieszczeniach o kontrolowanej temperaturze należy ustalić i udokumentować oprócz stabilności temperatury i jednorodności jej rozkładu, czas potrzebny do osiągnięcia warunków równowagi w odniesieniu do typowych zastosowań.
W laboratorium należy zapewnić oddzielne urządzenia chłodnicze do przechowywania: pożywek gotowych do użycia, odczynników, próbek do badań, szczepów i zawiesin drobnoustrojów.
Przed rozpoczęciem użytkowania autoklawów i sterylizatorów suchym gorącym powietrzem powinna być przeprowadzona walidacja. Należy badać przestrzenny rozkład temperatury dla wszystkich cykli pracy i wszystkich konfiguracji wsadu wykorzystywanego w praktyce. Zaleca się, oprócz bezpośredniego monitorowania temperatury sterylizacji, sprawdzanie pracy, w każdym cyklu za pomocą chemicznych lub biologicznych wskaźników, których wynik należy uznać jako potwierdzenie, że wsad został poddany procesowi, nie zaś jako potwierdzenie prawidłowego przebiegu cyklu sterylizacji.
Jest wymagane aby w laboratorium mikrobiologicznym wyposażenie do pomiarów objętości (automatyczne dozowniki, ręczne pipety automatyczne, pipety jednorazowego użytku), przed rozpoczęciem użytkowania było sprawdzone. Sprawdzenie należy przeprowadzać regularnie w celu zapewnienia, że przyrządy te działają zgodnie z wyspecyfikowanymi wymaganiami. Sprzęt jednorazowy do pomiaru objętości powinien pochodzić od firm posiadających uznany i odpowiedni system jakości. Dla takiego sprzętu należy wykonać sprawdzenie przed użytkowaniem, a następnie dokonywać sprawdzenia dokładności losowo. Natomiast sprawdzaniem przydatności należy objąć każdą partię przyrządów pochodzących od dostawcy, który nie posiada uznanego systemu jakości [pkt.6.2.6 w poz. 6].
W większości przypadków częstotliwość wzorcowania i sprawdzeń nie jest określona w dokumentach normatywnych i dlatego powinna być określona w każdym laboratorium, w zależności od typu urządzenia, liczby wykonywanych badań w laboratorium i zgodna z instrukcjami producenta. Konserwację wyposażenia należy wykonywać w określonych odstępach czasu biorąc pod uwagę częstotliwość użytkowania. Szczegółowe zapisy należy przechowywać. Przykłady dotyczące zakresu i częstotliwości wykonania konserwacji wyposażenia zawiera Załącznik F do dokumentu EA-4/10 [6] jak również norma ISO 7218 [12].
Wpływ jakości pożywek i odczynników na jakość analiz mikrobiologicznych
Niezawodność wyniku analizy mikrobiologicznej uzależniona jest od jakości stosowanych podłoży i odczynników. Ważnym elementem w pracy laboratorium mikrobiologicznego jest nadzór nad pożywkami, buforami i odczynnikami. Istnieją cztery kategorie pożywek stosowanych w laboratorium:
- gotowe do użycia, wytworzone w celach handlowych
- wymagające upłynnienia, uzupełnienia i rozlania
- przygotowane z dostępnych na rynku suchych granulatów
- przygotowane z pojedynczych składników.
Zgodnie z wymaganiami FP IX należy badać każdą serię podłoża stosowanego do badań aby potwierdzić, że stosowane podłoże jest właściwe do wykrywania drobnoustrojów w obecności produktu leczniczego [15]. Weryfikacja zgodności serii pożywki powinna być zakończona przed zwolnieniem analiz wykonywanych z użyciem tej pożywki. Termin przydatności do użycia pożywek przechowywanych w odpowiednich warunkach powinien być określony. Serie pożywek powinny być identyfikowalne.
Należy zapewnić aby wszystkie odczynniki, pożywki, rozcieńczalniki były właściwie oznakowane, a zapisy umożliwiały identyfikację osoby odpowiedzialnej za ich przygotowanie [6].
Szczepy testowe (kultury odniesienia) są wymagane do potwierdzenia parametrów akceptacji pożywek takich jak np.: odzysk lub przeżywalności pożądanych mikroorganizmów, hamowanie rozwoju lub eliminowania niepożądanych mikroorganizmów, właściwości różnicujących, oceny jałowości, pH, jak również do walidacji metod i stałej oceny jakości pracy. W laboratoriach mikrobiologicznych wytwórni farmaceutycznych, do oceny podłoży w określonych warunkach, stosowane są metody opisane w aktualnych farmakopeach. Mogą być również przydatne procedury opisane w normach ISO [16]. Do przeprowadzania tych badań konieczne jest zachowanie spójności pomiarowej. W celu wykazania spójności pomiarowej, laboratoria muszą korzystać ze szczepów odniesienia, drobnoustrojów uzyskanych bezpośrednio z uznanej kolekcji. Podczas przygotowywania szczepów macierzystych należy przeprowadzać sprawdzenie czystości i cech biochemicznych. W dokumencie EA-4/10 w Załączniku C zamieszczono schemat postępowania z kulturami odniesienia. Norma ISO 11133-1 zawiera diagram sposobu przygotowania szczepów roboczych ze szczepów macierzystych. Wszystkie etapy pozyskiwania szczepów odniesienia muszą być w pełni udokumentowane. Szczepy te należy przechowywać w postaci głęboko zamrożonych lub liofilizowanych porcji. Należy stosować stabilne zawiesiny szczepów testowych stosując takie techniki postępowania z materiałem siewnym aby zdolne do życia drobnoustroje używane do zaszczepienia, pochodziły z pasażu nie wyższego niż 5 pasaż macierzystej serii siewnej [15].
Pobieranie prób
Jednym z warunków, gwarantujących izolację i ilościowe oznaczenie tylko tych drobnoustrojów, które są obecne w próbkach, jest prawidłowe pobieranie prób. Czynności wykonywane przy pobraniu i opracowaniu próby powinny być opisane w procedurach uwzględniających zasady bezpieczeństwa i higieny obowiązujące w laboratorium. Próby materiałów wyjściowych, materiałów opakowaniowych, produktów pośrednich, produktów luzem i produktów końcowych są pobierane przez pracowników z zastosowaniem metod zatwierdzonych przez KJ. W pisemnych procedurach pobierania prób, należy wskazać osobę uprawnioną i określić: stosowane metody i używany sprzęt, wielkość pobieranych prób oraz środki ostrożności, jakie mają być zastosowane aby uniknąć zanieczyszczenia materiału lub jakiegokolwiek pogorszenia jego jakości. Należy również podać sposób podziału pobranej próby, określić rodzaj pojemników, jak również – jeśli ma to miejsce – specjalne środki ostrożności, związane z pobieraniem prób materiałów sterylnych lub szkodliwych dla zdrowia. Istotne są też warunki przechowywania urządzeń do pobierania prób po umyciu i wysterylizowaniu.
Próby do kontroli mikrobiologicznej środowiska badań i środowiska wytwarzania pobiera się zgodnie z planem poboru próbek opracowanym na podstawie oceny ryzyka otoczenia (pomieszczenia i powietrze) oraz analizy danych historycznych i analizy zagrożeń punktów krytycznych. Próby powinny być pobierane przez przeszkolony personel, w aseptyczny sposób przy użyciu wyjałowionego sprzętu. Pobranie próby do badań należy traktować jako część metody badawczej opisanej w odpowiedniej procedurze. W normie PN-EN ISO 6887-1 przedstawiono ogólne zasady przygotowania prób do badań [17]. Po przeprowadzonych badaniach próby – jak i wszystkie pozostałości po badaniach – należy utylizować zgodnie z procedurą uwzględniającą wymagania ustawy [18].
Literatura
- Eudralex The Rules Governing Medicinal Products in the European Union Volume 4 EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use.
- Rozporządzenie Ministra Zdrowia z dnia 1 października 2008 r. w sprawie Wymagań Dobrej Praktyki Wytwarzania (Dz. U. 2008, Nr 184, poz.1143 z późn. zm.).
- FDA U.S. Food and Drug Administration; Code of Federal Regulations Title 21 Food and Drug, Part 211 Current Good Manufacturing Practice for finished pharmaceuticals.
- J.F. Biron Koncepcja laboratorium mikrobiologicznego. Etapy kwalifikacji. Pharmaceutica. 2003, nr 21.
- Jadwiga Marczewska, Krystyna Mysłowska. GMP w przemyśle farmaceutycznym. LAB 3/2013
- EA-04/10. EA Europen co-operation for accreditation. Akredytacja laboratoriów mikrobiologicznych. Lipiec 2002 przegląd 02.
- Norma EN ISO/IEC 17025 Ogólne wymagania dotyczące kompetencji laboratoriów badawczych i wzorcujących
- Aneks 1 w Rozporządzenie Ministra Zdrowia z dnia 1 października 2008 r. w sprawie Wymagań Dobrej Praktyki Wytwarzania (Dz. U. 2008, Nr 184, poz.1143 z późn. zm.).
- Raport komisji SFSTP pod przewodnictwem: J.P. Farcy, V. le Juez Dobra praktyka mikrobiologiczna w laboratorium. Pharmaceutica. 2002, nr 19.
- Mikrobiologia Techniczna. Red. Nauk.: Z. Libudzisz, K. Kowal, Z. Żakowska. Wydawnictwo PWN. 2008
- Dyrektywa 2000/54/WE Parlamentu Europejskiego i Rady z dnia 18 września 2000 r. w sprawie ochrony pracowników przed ryzykiem związanym z narażeniem na działanie czynników biologicznych w miejscu pracy ( siódma dyrektywa szczegółowa w rozumieniu art. 16 ust.1 dyrektywy 89/391/EWG).
- Norma PN-EN ISO 7218:2008 Mikrobiologia żywności i pasz Wymagania ogólne i zasady badań mikrobiologicznych.
- Rozporządzenie MZ z dnia 22 kwietnia 2005 r. w sprawie szkodliwych czynników biologicznych dla zdrowia w środowisku pracy oraz ochrony zdrowia pracowników zawodowo narażonych na te czynniki. (Dz. U. 2005 Nr 81, poz. 716 z późniejszymi zmianami).
- Tomasz Zaręba. Mikrobiologia farmaceutyczna. III Konferencja naukowo-szkoleniowa, 23-25 maja 2012. Wieliczka.
- Farmakopea Polska. Wydanie IX (2011), Tom I, monografia 07/2010:20612
- Norma PKN-CEN ISO/TS 11133-1 Mikrobiologia żywności i pasz. Wytyczne dotyczące przygotowania i wytwarzania pożywek. Część 1: Ogólne wytyczne dotyczące kontroli jakości pożywek przygotowanych w laboratorium
- Norma PN-EN ISO 6887-1 lipiec 2000. Mikrobiologia żywności i pasz. Przygotowanie próbek, zawiesiny wyjściowej i rozcieńczeń dziesięciokrotnych do badań mikrobiologicznych.
- Ustawa z dnia 14 grudnia 2012 o odpadach (Dz. U. z dnia 8 stycznia 2013, poz. 21).
*Dr Jadwiga Marczewska; jmarczewska@interia.pl(link sends e-mail); mgr Krystyna Mysłowska; krystynamyslowska@gmail.com(link sends e-mail)
*Dr Jadwiga Marczewska; jmarczewska@interia.pl(link sends e-mail); mgr Krystyna Mysłowska; krystynamyslowska@gmail.com